

王永飞研究组合作揭示胞质Ca2+信号级联放大的分子机制
气孔是由一对特化表皮细胞,即保卫细胞,围绕而成的孔型结构,是植物与外界环境进行气体交换和水分蒸腾的“门户”。植物通过精细调控气孔开闭,以平衡光合效率与水分流失,从而优化生长发育并应对逆境胁迫。当遭遇干旱胁迫时,植物通过合成和积累脱落酸(ABA)触发气孔关闭,这是植物抵御干旱的核心策略。在这一气孔开度调控过程中,ABA引发保卫细胞内钙离子(Ca2+)浓度的周期性振荡,即Ca2+信号,该Ca2+信号是触发气孔关闭的关键上游事件。然而,气孔保卫细胞如何精确编码这一特异性的Ca2+信号,其底层的分子机制是怎样的,一直是植物学领域的核心科学问题。
2026年6月30日,国际学术期刊 PNAS在线发表了中国科学院分子植物科学卓越创新中心王永飞研究组合作完成的题为 “CPKs are involved in Ca2+ signaling encoding by enhancing OST1-initiated Ca2+ influx for ABA-induced stomatal closure in Arabidopsis”的研究论文。该研究深入揭示了Ca2+依赖型蛋白激酶CPK3/8/10介导的正反馈环路在放大和维持Ca2+信号中的关键作用。
王永飞研究团队此前的研究已经表明,ABA激活的拟南芥气孔保卫细胞质膜Ca2+通道主要由CNGC5/6/9/12四个蛋白构成,而不依赖Ca2+的蛋白激酶 OST1通过磷酸化这些通道蛋白的N端上的一个保守丝氨酸位点来激活其Ca2+通道活性,引发胞外Ca2+内流和胞内Ca2+信号(Tan and Yang et al., Plant Cell 2023; Yang et al., Plant Cell 2024)。这一发现回答了“Ca2+信号如何被诱发”的问题,但Ca2+信号被放大和编码的机理依然不清楚。
研究人员通过电生理筛选发现,CPK3/8/10均能以Ca2+依赖的方式直接激活 CNGC5/6/9/12 ,其中CPK3为主效组分。同时,在cpk3/8/10三突变体中,ABA诱导的气孔关闭及Ca2+振荡均受到显著抑制。研究人员进一步利用蛋白质谱、体外磷酸化及膜片钳技术,通过大量的体内外实验,鉴定出了CPK3磷酸化CNGCs的主效靶点,该位点位于其C自由端,与位于N自由端的OST1的磷酸化位点不同。这一发现预示着一种双重调控机制的存在。
进一步的深入研究发现,OST1和CPK3的磷酸化位点同时被磷酸化对CNGC的激活作用明显强于单个位点的磷酸化,单个位点的磷酸化所产生的激活作用可以被另一个位点的去磷酸化所抑制,且这两个位点同时去磷酸化强力抑制CNGC的活性。在气孔保卫细胞内,响应ABA和干旱刺激,OST1首先激活CNGCs以起始胞外Ca2+内流和胞内Ca2+升高,Ca2+的升高激活CPK3,最后CPK3通过磷酸化CNGCs的C端提高其Ca2+通道活性。
基于这些实验结果,研究团队提出了一个“OST1启动-CPK3放大”的Ca2+信号编码模型。在信号启动阶段,OST1首先磷酸化CNGCs的N端,触发初始的Ca2+内流;胞内Ca2+的升高迅速诱发Ca2+与CPK3结合并激活其激酶活性;被激活的CPK3通过磷酸化CNGCs的C端进一步显著增强其Ca2+通道活性,导致更大规模的Ca2+内流。特别值得注意的是,胞内Ca2+升高、CPK3的激活和CPK3对CNGCs的激活形成一个正反馈环路,迅速放大胞内Ca2+信号,最终有效关闭气孔。该正反馈循环机制还控制胞内Ca2+震荡的模式,即参与Ca2+信号编码。这一发现不仅深化了人们对植物细胞Ca2+信号编码机制的理解,也为未来通过基因编辑技术改良作物抗旱性提供了新的分子靶点。
分子植物卓越中心王永飞研究组副研究员谭艳秋、博士研究生任映玥和博士后杨阳博士为该论文的共同第一作者,王永飞研究员和深圳南方科技大学王鹏程教授为共同通讯作者。赵杨研究组和张鹏研究组(结构生物学)参与了部分研究工作。本研究得到了中国科学院先导专项(B类)、国家自然科学基金、中国博士后科学基金和上海市“超级博士后”激励计划等项目的资助。
论文链接:https://doi.org/10.1073/pnas.2537976123
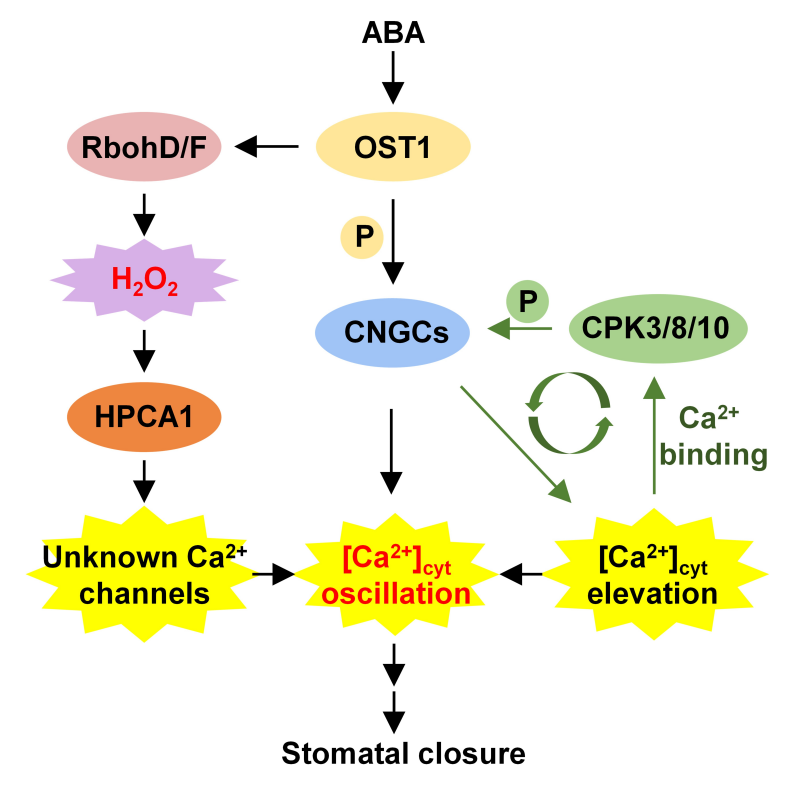
图1. 拟南芥气孔保卫细胞中,ABA特异性Ca2+信号编码机制示意图。
响应干旱和ABA刺激,OST1磷酸化CNGC5/6/9/12 N端一个保守丝氨酸位点,激活这些CNGC的Ca2+通道活性,引发胞外Ca2+内流和胞质Ca2+升高。这第一波次的胞质Ca2+升高引起了Ca2+与靠近质膜分布的CPK3/8/10的结合,并因此激活这些CPK的激酶活性。这三个CPK进一步磷酸化位于CNGCs C端的一个保守丝氨酸位点,提高其Ca2+通道活性,引起更大规模的胞外Ca2+内流和更强的胞内Ca2+升高。这样,胞内Ca2+升高、Ca2+与CPKs的结合,以及CPKs对CNGCs的磷酸化形成一个正反馈环路,以正循环的方式迅速放大胞质Ca2+信号,最终加速诱导气孔的关闭。这一机制不仅仅简单地负责胞质Ca2+的升高,还参与Ca2+震荡模式的编码。其中,活性自由基(ROS)通过ROS的受体HPCA1激活一种未知的质膜Ca2+通道,参与胞质Ca2+信号的编码和调控(Tan et al., PNAS 2026)。

Copyright © 2002-
中国科学院分子植物科学卓越创新中心 版权所有
地址:中国上海枫林路300号(200032)
电话:86-21-54924000
传真:86-21-54924015
Email: webmaster@cemps.ac.cn